Hartree-Fock-Näherung

Karl-Wilhelm Steinfieber
Atom- und Molekülphysik, Hartree-Fock-Verfahren, quantenmechanisches Näherungsverfahren zur Bestimmung der Wellenfunktionen und Energieniveaus bei mehreren identischen Fermionen, insbesondere Elektronen im Atom oder im Festkörper. Ursprünglich wurde das Hartree-Fock-Verfahren zur genaueren theoretischen Behandlung der Struktur der Atomhülle entwickelt. Es wurde von Hartree zunächst ohne, von Fock dann mit Berücksichtigung des Elektronenspins und der Austauschwechselwirkung formuliert. Nach dem Modell unabhängiger Teilchen besteht die Grundannahme in der Existenz von Einelektronen-Wellenfunktionen, wobei die entsprechenden Zustände unter Beachtung des Pauli-Prinzips besetzt werden. Dabei wird die Wellenfunktion eines beliebig herausgegriffenen Elektrons als Lösung einer Schrödinger-Gleichung bestimmt, in der Wechselwirkungen mit den anderen Elektronen durch ein mittleres Potential approximiert werden. Diese Grundidee, nämlich der Ersatz der Wechselwirkung zwischen den Elektronen durch ein Einteilchenpotential, ist schon im Rahmen der älteren Quantentheorie erfolgreich erprobt worden (Abschirmung).
Die
Gesamtfunktion wird als Produkt (Hartree) y(1, ..., N) = j1(1)j2(2) ...jN(N) bzw. als antisymmetrisiertes Produkt (Fock)
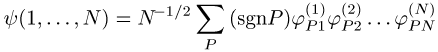
geschrieben, die Summation läuft über alle Permutationen P(1, 2, ..., N) = (Pl, P2, ..., PN) der Zahlen 1 ... N, sgn P ist das Vorzeichen der Permutation P, N ist die Teilchenzahl. Dabei kann die antisymmetrische Wellenfunktion auch in der Form der Slater-Determinante
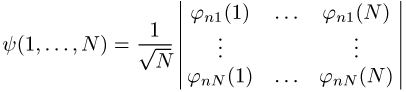
formuliert werden. Im Argument der Einteilchenwellenfunktionen
sind der Ortsvektor rk und die
Spinvariable sk zusammenfassend
mit k bezeichnet. Die Spinvariable sk kann nur die Werte ±1 / 2 annehmen.
Bei Vernachlässigung magnetischer Wechselwirkungen können die ![]() als Produkte aus je einer nur von der
Ortskoordinate rk abhängigen und
einer nur von der Spinvariablen sk
abhängigen Funktion angesetzt werden:
als Produkte aus je einer nur von der
Ortskoordinate rk abhängigen und
einer nur von der Spinvariablen sk
abhängigen Funktion angesetzt werden: ![]() . Für die
Spinfunktionen
. Für die
Spinfunktionen ![]() könnte man etwa eine Darstellung durch ein
Kronecker-Symbol
könnte man etwa eine Darstellung durch ein
Kronecker-Symbol ![]() wählen. Bestimmungsgleichungen für die
Einteilchenwellenfunktionen erhält man aus der Extremaleigenschaft der Energie
wählen. Bestimmungsgleichungen für die
Einteilchenwellenfunktionen erhält man aus der Extremaleigenschaft der Energie ![]() mit dem Vielelektronen-Hamilton-Operator
mit dem Vielelektronen-Hamilton-Operator
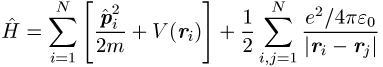
eines nichtrelativistischen Elektronensystems, wobei die ![]() variiert werden. Mit dem gegenüber
Teilchenzahlvertauschungen antisymmetrischen Hartree-Fock-Ansatz erhält man das
folgende nichtlineare System von Integrodifferentialgleichungen zur Berechnung
der Einteilchenwellenfunktionen
variiert werden. Mit dem gegenüber
Teilchenzahlvertauschungen antisymmetrischen Hartree-Fock-Ansatz erhält man das
folgende nichtlineare System von Integrodifferentialgleichungen zur Berechnung
der Einteilchenwellenfunktionen ![]() :
:
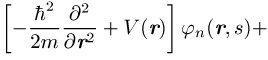
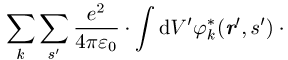
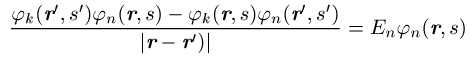
Die Integration erstreckt sich über den ganzen Raum und die s¢-Summe über die Werte ±1 / 2 der Spinvariablen. Die k-Summe umfasst alle besetzten Einteilchenzustände. Berücksichtigt man, dass die Summation über s¢ für ein Produkt jn(r¢, s¢)jm(r¢, s¢) stets null liefert, wenn die Spins beider Zustände nicht übereinstimmen, so ergibt sich folgendes: Das effektive Potential in den Gleichungen des Hartree-Fock-Verfahrens besteht zunächst wie beim Hartree-Verfahren aus dem Potential V(r), das auf jedes einzelne Elektron wirkt und das bei einzelnen Atomen gleich dem Coulomb-Potential -Ze2 / 4pe0r des Z-fach geladenen Kerns, bei Kristallen gleich der gitterperiodischen Überlagerung der Potentiale aller Ionenrümpfe ist, und dem Potential, das alle anderen Elektronen im Mittel erzeugen. Der Beitrag der Elektronen, deren Spinrichtung mit der des Zustandes jn übereinstimmt, zum effektiven Potential wird durch den Austauschterm korrigiert. Der Austauschterm beruht auf der Korrelation der Elektronen infolge der Antisymmetrie der Wellenfunktion; er tritt bei dem einfachen Hartreeschen Produktansatz nicht auf und berücksichtigt ein zusätzliches gegenseitiges Ausweichen der Elektronen gleichen Spins. Nach dem Koopmansschen Theorem kann E als diejenige Energie interpretiert werden, die man aufwenden muss, um ein im Zustand jn befindliches Elektron aus dem System zu entfernen.
Bei Anwendung auf Atome werden die Einteilchenwellenfunktionen
![]() im Atom durch die Hauptquantenzahl n, die Drehimpulsquantenzahlen l
und ml sowie die
Spinquantenzahl ms charakterisiert.
im Atom durch die Hauptquantenzahl n, die Drehimpulsquantenzahlen l
und ml sowie die
Spinquantenzahl ms charakterisiert.
Die über die Berechnung einer Elektronenkonfiguration bei nichtabgeschlossenen Elektronenschalen bzw. Unterschalen auftretende Feinstruktur kann nicht mit einer einzigen Slater-Determinante erfasst werden.
Das dem Austauschterm entsprechende Austauschpotential ist nichtlokal und erschwert die Lösung des Hartree-Fock-Verfahrens beträchtlich. In Festkörpern mit ihren vielen Elektronen wird es daher stets lokal approximiert (Slater-Methode). In dem so entstehenden Potential können auch über das Hartree-Fock-Verfahren hinausgehende Korrelationseffekte berücksichtigt werden (Dichtefunktionaltheorie).
Für ein freies Elektronengas mit einem homogenen »Untergrund«
positiver Ladungen geben sich in Hartreescher Näherung, also bei völliger
Vernachlässigung des Austauschterms, die Energien zu ![]() , während das
Hartree-Fock-Verfahren einen Zusatzterm liefert, der an der Fermi-Fläche eine
logarithmisch divergente Ableitung hat, d.h. die Fermi-Geschwindigkeit
divergiert in Hartree-Fock-Näherung, und die Zustandsdichte N(EF) an der Fermi-Energie
verschwindet. Dieses anomale Verhalten tritt bei lokaler Approximation des
Austauschterms (Slater-Methode) nicht auf. Bei einfachen Metallen ist dann der
Einfluss des Austauschpotentials auf die Energien E(k) wegen der räumlich nahezu homogenen
Valenzelektronendichte gering. Bei Halbleitern und Übergangsmetallen ergibt die
Berücksichtigung des Austauschpotentials jedoch eine bedeutende Verbesserung
des Energiespektrums, verglichen mit Hartree-Rechnungen. Konkrete Rechnungen
mit dem Hartree-Fock-Verfahren sind sehr aufwendig, auf iterativem Weg wird
eine selbstkonsistente Lösung bestimmt (self-consistent field).
, während das
Hartree-Fock-Verfahren einen Zusatzterm liefert, der an der Fermi-Fläche eine
logarithmisch divergente Ableitung hat, d.h. die Fermi-Geschwindigkeit
divergiert in Hartree-Fock-Näherung, und die Zustandsdichte N(EF) an der Fermi-Energie
verschwindet. Dieses anomale Verhalten tritt bei lokaler Approximation des
Austauschterms (Slater-Methode) nicht auf. Bei einfachen Metallen ist dann der
Einfluss des Austauschpotentials auf die Energien E(k) wegen der räumlich nahezu homogenen
Valenzelektronendichte gering. Bei Halbleitern und Übergangsmetallen ergibt die
Berücksichtigung des Austauschpotentials jedoch eine bedeutende Verbesserung
des Energiespektrums, verglichen mit Hartree-Rechnungen. Konkrete Rechnungen
mit dem Hartree-Fock-Verfahren sind sehr aufwendig, auf iterativem Weg wird
eine selbstkonsistente Lösung bestimmt (self-consistent field).
Techniklexikon.net
Das freie Technik-Lexikon. Fundierte Informationen zu allen Fachgebieten der Ingenieurwissenschaften, für Wissenschaftler, Studenten, Praktiker & alle Interessierten. Professionell dargeboten und kostenlos zugängig.
Techniklexikon
Modernes Studium der Physik sollte allen zugängig gemacht werden.